Атипичная эволюция роландической эпилепсии синдром псевдо леннокса

Доброкачественную роландическую эпилепсию относят к генетическим неврологическим заболеваниям. У пациентов отмечается повышенная возбудимость височной коры больших полушарий, что обуславливает возникновение симптомов. Основные клинические проявления — судорожные приступы, имеющие локальный или генерализованный характер. Для подтверждения диагноза необходимо проведение ЭЭГ. Как правило болезнь самостоятельно проходит у детей при взрослении.
Этиология заболевания
Причина развития болезни неизвестна. У 40-70% пациентов отмечается наследование патологии. Оно носит мозаичный характер, т. е. встречается не у всех родственников. На сегодняшний день в неврологии считают, что за развитие роландической эпилепсии ответственны два гена, определяющих работу нейронов коры больших полушарий.
Основная теория объясняет возникновение болезни незрелостью корковых отделов головного мозга, преимущественно центральной части височной доли. Эпилепсия связана с возрастом пациента и возникает преимущественно у детей. Кора больших полушарий в детском возрасте отличается рядом особенностей:
- большое количество возбуждающих нейромедиаторов, превосходящих по количеству тормозящие молекулы;
- µ-аминомасляная кислота и рецепторы к ней выявляются в небольшом количестве;
- большая часть синапсов носит возбуждающий характер;
- эпилептогенные области ЦНС (гиппокамп, лимбическая система) имеют высокий уровень активности.
Указанные факторы по мере взросления ребенка устраняются, что приводит к снижению частоты приступов роландической эпилепсии и полному выздоровлению. У взрослых заболевание не встречается.
Клинические проявления
Парциальные приступы без утраты сознания — основной симптом роландической эпилепсии при ее классическом течении. У больных до возникновения эпиприступа отмечается сенсорная аура: чувство покалывания или онемения в области лица, губ, языка или глотки. После этого появляется моторный компонент в виде тонических, клонических судорог или их сочетания.
Приступы роландической эпилепсии бывают двух форм: фарингооральный и гемифациальный.
- При фарингооральном судороги возникают в мышечных группах глотки, гортани, языка и губ. Они носят односторонний характер и приводят к изменению речи, а также к повышенному слюноотделению. Во время эпиприступа ребенок может издавать неприятные звуки.
- Гемифациальный сопровождается односторонним сокращением мимической и жевательной мускулатуры.
У большинства детей приступы эпилепсии возникают перед сном или сразу после пробуждения. Специалисты считают, что это связано с особенностями работы головного мозга в ночное время. Припадки днем встречаются редко. У 15-25% детей возможно распространение судорог на руку, а у 5% — на ногу.
На фоне парциальных эпиприступов возможно развитие вторично-генерализованных судорожных припадков, которые сопровождаются тоническими и клоническими сокращениями мускулатуры всего тела и потерей сознания.
При атипичной роландической эпилепсии возникают полиморфные приступы. Они могут носить характер фокальных моторных, атонических припадков, атипичных абсансов, генерализованных судорог и эпилептического статуса.
Диагностические мероприятия
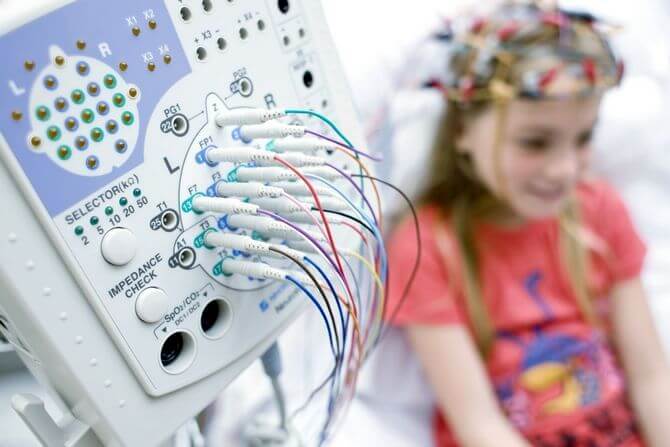
Обследование больных проводится по определенному алгоритму:
- Беседа с родителями ребенка и с самим больным, в ходе которой выясняется давность появления приступов, их характер и особенности возникновения — время появления, длительность, сохранение сознания и др. Важно установить подробности течения беременности и родов, так как органические повреждения головного мозга в эти периоды могут быть причиной вторичной эпилепсии.
- Неврологический осмотр направлен на выявление симптомов поражений центральной нервной системы. Специалист может выявить нарушения чувствительности, мышечной силы и другие симптомы, свидетельствующие о поражении структур головного мозга.
- ЭЭГ (электроэнцефалография) — «золотой стандарт» диагностики роландической эпилепсии. Больным проводится ЭЭГ-мониторинг в течение ночи или полисомнография, позволяющие выявить изменения активности отдельных участков головного мозга.
- Для исключения органических повреждений ЦНС (внутримозговых опухолей, изменений, связанных с черепно-мозговыми травмами, абсцессов, менингитов) проводится магнитно-резонансная томография.
На ЭЭГ отмечаются волны и пики острой формы, говорящие о высокоамплитудной активности. После них могут фиксироваться медленные волны. Два указанных изменения объединяются под названием «роландический комплекс», который выявляется у 80-90% больных. При повторных проведениях электроэнцефалографии картина меняется, что свойственно для данной формы эпилепсии.
Отличия от других заболеваний
При проведении дифференциальной диагностики необходимо исключить органические поражения головного мозга, ночную эпилепсию и синдром псевдо-Леннокса.
Внутримозговые опухоли, изменения при черепно-мозговых травмах, менингите и энцефалите могут привести к появлению эпилептических приступов. Для подобных заболеваний характерно наличие стойкой неврологической симптоматики (снижение мышечной силы, появление парастезий, нарушения зрения и др.). При роландической эпилепсии на ЭЭГ всегда присутствует нормальная основная активность.
У 3-7% больных возможно развитие синдрома псевдо-Леннокса. В этом случае помимо классических приступов возникают абсансы, нарушения памяти и мышления и другие симптомы, что приводит к трудностям в диагностике. При синдроме псевдо-Леннокса при проведении электроэнцефалографии возникают медленные комплексы или диффузная пиковая активность.
Ночная эпилепсия со сложными парциальными приступами — патология, сопровождающаяся схожими симптомами в ночное время. Основное отличие связано с тем, что больные теряют сознание, а на ЭЭГ имеются изменения в активности лобных отделов головного мозга.
Интерпретировать результаты проводимых исследований должен только врач. Попытки самостоятельной постановки диагноза, а также изменение назначенного специалистом лечения может стать причиной прогрессирования патологии или развития побочных эффектов от применения медикаментов.
Подходы к лечению

Однозначного ответа о том, лечить или нет роландическую эпилепсию нет. Патология является доброкачественной и проходит самостоятельно без использования каких-либо медикаментозных средств. Исходя из этого, ряд специалистов отказывается от назначения противоэпилептических препаратов.
Большая часть неврологов считает, что использование лекарственных средств все же необходимо. У больных есть риск развития атипичной фокальной эпилепсии, характеризующейся частыми приступами без тенденции к самостоятельному выздоровлению. Кроме того, под диагнозом роландических эпиприступов могут скрываться другие типы эпилепсии в случае неправильной диагностики. В отсутствии противоэпилептических препаратов, данные патологии способны быстро прогрессировать.
В лечении используют медикаменты на основе вальпроевой кислоты — Депакин, Энкорат хроно и др. Их применяют в виде монотерапии, т. е. не комбинируют с другими средствами. Если вальпроевая кислота не эффективна, то ее заменяют на Леветирацетам. При возрасте ребенка старше 7 лет возможно использование Карбамазепина.
Помимо указанных лекарственных препаратов, специалисты не рекомендуют использовать другие медикаменты с противосудорожной активностью или обладающие психостимулирующим действием. Это может стать причиной развития побочных эффектов, затрудняющих терапию заболевания.
Современные научные исследования показывают, что большое значение в профилактике эпилепсии имеет питание. Положительный эффект наблюдается при переходе на кетогенную диету. Рацион подбирается индивидуально, в зависимости от возраста ребенка, его веса, а также уровня физической и интеллектуальной нагрузки в течение дня. Питание при кетодиете основывается на повышенном потреблении жиров, которые в организме трансформируются в кетоновые тела, улучшающие работу головного мозга. Следует отметить, что во время перехода на диету необходимо постоянно находится под присмотром врача. При этом больному ежедневно определяют уровень кетоновых тел в крови, оценивая эффективность диеты.
Правила первой помощи
Эпилептические приступы у детей возникают в вечернее или утреннее время, так как связаны со сном. Родителям необходимо знать, как оказать первую помощь.
При появлении признаков ауры – слабости, ощущение больным неприятного запаха, его следует уложить на мягкую кровать. Это позволяет предупредить травмы при развитии вторично-генерализованного приступа.
Во время эпиприпадка маленького ребенка нужно приподнять, крепко удерживая его за тело и голову. Это необходимо для предупреждения развития травм в результате ударов о твердые предметы: стену, элементы мебели и др. Ни в коем случае не стоит пытаться удержать больного крепко, сжав ноги и руки. В этом случае имеется риск травматизации мышц и связочного аппарата.
Вызвать скорую медицинскую помощь нужно в том случае, когда приступ носит генерализованный характер и сопровождается потерей сознания.
Предупреждение заболевания

Первичная профилактика патологии отсутствует.
Для профилактики судорожных приступов выделяют ряд рекомендаций:
- Обеспечить ребенку постоянный режим дня с достаточным временем на отдых. Одна из причин развития эпиприпадка — нарушение сна в виде его малой продолжительности.
- Спать больной должен в общей с родителями комнате, так как его нежелательно оставлять одного. Если это невозможно, следует использовать радионяню. Родители, находящиеся рядом с ребенком, могут своевременно заметить ауру и оказать необходимую первую помощь.
- За 1-2 часа до сна необходимо исключить игры за компьютером, смартфоном, а также любую нагрузку, стимулирующую работу головного мозга. Кроме того, не следует плотно кормить детей перед сном.
Прогноз
Прогноз при роландической эпилепсии благоприятный. В возрасте 15 лет у 90% больных наблюдается полное выздоровление без каких-либо остаточных симптомов. У 1-3% подростков эпизодические эпилептические приступы сохраняются, однако, их число не увеличивается.
Неблагоприятный прогноз с прогрессированием патологии возможен в тех случаях, когда первые эпизоды болезни возникают до 4 или после 10 лет жизни. При этом у детей имеется склонность к вторично-генерализованным эпиприпадкам, сопровождающимися потерей сознания. Эффективность медикаментов при этом снижена.
Рецидивы возможны спустя несколько лет бессимптомного периода. Если заболевание сохраняется во взрослом возрасте, то приступы носят единичный характер. Больной может не придавать им значение, так как они возникают в ночное время и могут оставаться незамеченными.
Медицинское наблюдение и применение противоэпилептических средств позволяет предупредить прогрессирование болезни и развитие осложнений. При первых симптомах эпилепсии следует обращаться за профессиональной медицинской помощью к врачу.
Статья по теме: Эпилепсия у детей
Источник

Синдром Леннокса-Гасто — отдельная форма эпилепсии детского возраста, характеризующаяся наличием полиморфных пароксизмов (миоклонических, атонических, тонических и абсансов) и задержкой нейро-психического развития. Может иметь криптогенный характер или выступать синдромом других патологических состояний (церебральных аномалий, генетических обменных заболеваний, перинатальной патологии). Синдром Леннокса-Гасто диагностируется по типичной вариативной картине эпиприступов и характерному паттерну электроэнцефалограммы. Дополнительно проводится МРТ и КТ головного мозга. Антиконвульсантная терапия синдрома малоэффективна, проводится поиск альтернативных методов лечения. Прогноз вариабельный, но в большинстве случаев неблагоприятный.
Общие сведения
Синдром Леннокса-Гасто (СЛГ) — вариант эпилепсии детского возраста, для которого характерно сочетание атонических, миоклонических, тонических эпиприступов и атипичных абсансов, медленный островолновой паттерн ЭЭГ. В 1950 г. СЛГ был выделен в качестве отдельного эпилептического синдрома, а в 1964-1966 гг. неврологическое сообщество признала его самостоятельной нозологической формой. Синдром Леннокса-Гасто по различным данным составляет от 3% до 10% всех случаев детской эпилепсии. Его распространенность колеблется в пределах 1-2,8 случаев на 10 тыс. Несколько чаще встречается у мальчиков. Типичный возраст начала заболевания от 2 до 5 лет, реже — 6-8 лет. Сегодня СЛГ является тяжелым заболеванием с прогрессирующим течением, эффективное лечение которого пока является предметом надежд многих специалистов в области детской неврологии и эпилептологии.

Синдром Леннокса-Гасто
Причины синдрома Леннокса-Гасто
Синдром Леннокса-Гасто относится к заболеваниям, этиологические факторы которых пока точно не установлены. Известно, что во многих случаях синдром носит симптоматический характер и формируется на фоне генетической патологии, последствий различных неблагоприятных факторов, действующих в перинатальном периоде и на 1-ом году жизни. Однако в большинстве случаев морфологический субстрат заболевания остается не выявленным. К этиофакторам, способным спровоцировать развитие СЛГ, относят гипоксию плода, внутриутробные инфекции (краснуху, цитомегалию, герпес, токсоплазмоз), родовые травмы новорожденных (в первую очередь внутричерепные), недоношенность, асфиксию новорожденных, тяжелые инфекционные заболевания постнатального периода (менингит, энцефалит), аномалии развития головного мозга (гидроцефалию, кортикальную дисплазию, гипоплазию мозолистого тела и др.), метаболические нарушения с поражением ЦНС, отдельные генетические заболевания (например, туберозный склероз).
В 25-40% случаев синдром Леннокса-Гасто возникает у детей с отягощенным по эпилепсии семейным анамнезом. Кроме того, существует гипотеза об этиологической роли иммунных нарушений, в т. ч. возникающих вследствие вакцинации. Примерно в 30% случаев СЛГ является следствием эволюции синдрома Веста. Когда синдром Леннокса-Гасто манифестирует на фоне полного благополучия в здоровье ребенка и отсутствия в его анамнезе вышеперечисленных факторов, говорят о криптогенной (не имеющей вероятной причины) форме заболевания. Криптогенный вариант СЛГ встречается в 10-20% случаев и отличается более благоприятным течением.
Симптомы синдром Леннокса-Гасто
Симптоматический синдром Леннокса-Гасто, как правило, дебютирует на фоне уже имеющегося отставания в умственном и психическом развитии. При криптогенной форме развитие ребенка на момент манифестации синдрома соответствует норме. СЛГ отличается большой вариативностью приступов, их различной продолжительностью и частотой.
Атонические пароксизмы обусловлены кратковременной утратой мышечного тонуса. При их генерализованном характере происходит падение ребенка, т. н. «дроп-атака». Локальные пароксизмы могут иметь вид внезапного подгибания коленей, выпадения предметов из рук, кивков головой и т. п. Отличительной чертой атонических эпизодов при СЛГ является их молниеносность и кратковременность (до 5 сек.). Генерализованные атонические пароксизмы СЛГ требуют дифференцировки от приступов миоклонически-астатической эпилепсии, обмороков, ОНМК.
Миоклонические пароксизмы представляют собой локальные мышечные подергивания. Чаще охватывают мышцы-сгибатели проксимальных отделов рук, при распространении на нижние конечности происходит падение. Характеризуются симметричным серийным возникновением в обеих конечностях и стереотипностью. Нуждаются в дифференцировке с миоклониями при клещевом энцефалите и токсических поражениях ЦНС; миоклонусом неэпилептического характера, для которого типичны нерегулярные асимметричные миоклонии, возникающие в ответ на различные сенсорные раздражители (звук, свет, прикосновение) и не сопровождающиеся изменениями ЭЭГ.
Тонические пароксизмы СЛГ часто возникают в период сна и отличаются своей кратковременностью (средняя длительность 10 сек.). Сопровождаются отключением сознания. Могут иметь генерализованный характер или проявляться в виде тонического напряжения отдельных мышечных групп (заднешейных, спинных, мышц брюшного пресса, плечевого пояса и пр.). Тонические пароксизмы сопровождаются тахикардией, цианозом лица, слезотечением, апноэ, гиперсаливацией. Минимальные локальные пароксизмы тонического характера иногда с трудом можно отдифференцировать от зевоты или потягивания.
Атипичные абсансы связаны с частичным нарушением сознания. Проявляются временным «оцепенением», отсутствием любой двигательной активности. При малой продолжительности абсансы зачастую не распознаются окружающими ребенка людьми. При СЛГ абсансы могут сопровождаться мышечной гипотонией (атонические абсансы) и гипертонусом мышц спины (ретропульсивные абсансы). Чаще, чем другие виды эпилепсии, синдром Леннокса-Гасто сопровождается статусом абсансов — непрерывно следующими друг за другом абсансами. Такой бессудорожный эпистатус обычно возникает при пробуждении, может длиться несколько часов и дней.
Задержка психомоторного развития (ЗПР) отмечается почти во всех случаях СЛГ. Ее выраженность зависит от формы синдрома (криптогенная или симптоматическая), характера фоновой патологии ЦНС, тяжести и частоты эпилептических пароксизмов. Как правило, на первый план выходят проблемы с обучением владению новыми навыками и с усвоением новой информации. Зачастую наблюдается агрессивность, гиперактивность, эмоциональная нестабильность, характерные для аутизма особенности характера. Около 50% подростков, имеющих синдром Леннокса-Гасто, не владеют навыками самообслуживания. Еще 25% социально и эмоционально дезадаптированы по причине выраженной олигофрении. Особенности поведения и характера не дают возможность нормально адаптироваться в социуме даже тем пациентам, у которых олигофрения имеет легкую степень выраженности. Нормальная социальная адаптация наблюдается лишь в 15% случаев.
Диагностика синдрома Леннокса-Гасто
Синдром Леннокса-Гасто устанавливается на основании типичной клинической картины, состоящей из полиморфных эпиприступов и симптомов отставания нейро-психического развития. Учитывается также возраст начала пароксизмов и семейный эпилептический анамнез. Большую диагностическую роль играет электроэнцефалография. Межприступная (интериктальная) ЭЭГ в бодрствующем состоянии регистрирует плохую структурированность и замедленность основного ритма. ЭЭГ-паттерн имеет картину гипсаритмии с большим количеством спайков различной амплитуды. Наиболее высокие пики регистрируются в лобной области. ЭЭГ-паттерн в период приступов зависит от их формы.
Методы нейровизуализации (МРТ и КТ головного мозга) выявляют преимущественно неспецифичные патологические изменения: внутреннюю гидроцефалию, атрофию подкорковых областей и корковых структур преимущественно лобной зоны, гипоплазию лобных долей. Попытки проанализировать при помощи ПЭТ головного мозга степень утилизации глюкозы церебральными тканями дали противоречивые сведенья: в одних случаях были выявлены зоны гиперметаболизма, в других — гипометаболизма; у части пациентов метаболизм глюкозы был в пределах нормы.
По причине большой вариативности пароксизмов, синдром Леннокса-Гасто следует дифференцировать с целым рядом других форм эпилепсии, дебютирующих в детском возрасте: с миоклонической эпилепсией, доброкачественной роландической эпилепсией, синдромом Веста, детской абсансной эпилепсией, дисметаболической эпилепсией при болезни Гоше, Краббе, Ниманна-Пика и др.
Лечение синдрома Леннокса-Гасто
Терапия проводится противоэпилептическими средствами. Применяются вальпроевая к-та, этосуксимид, карбамазепин, ламотриджин и др. В большинстве случаев проводится комбинированное лечение одним из указанных фармпрепаратов и вальпроатом натрия. Однако до 90% случаев синдрома Леннокса-Гасто являются резистентными к антиконвульсантной терапии. В связи с этим основной целью лечения является уменьшение числа эпиприступов и улучшение качества жизни ребенка и его семьи в межпароксизмальный период.
Неврологами и эпилептологами ведется поиск новых способов терапии. Доказанной является положительная роль кетогенной диеты, заключающейся в резком ограничении употребления углеводов и повышении содержания жиров в пище. Рядом клиницистов отмечен положительный эффект лечения синдрома Леннокса-Гасто большими дозировками иммуноглобулина. Наблюдалась эффективность применения АКТГ и глюкокортикоидов. В случаях, когда синдром Леннокса-Гасто сопровождается частыми и тяжелыми эпипароксизмами с падением и угрозой травматизации ребенка, совместно с нейрохирургом может быть рассмотрен вопрос о проведении хирургической операции рассечения мозолистого тела — каллозотомии. Подобное вмешательство не избавляет пациентов от приступов, но существенно уменьшает их интенсивность.
К новым способам лечения относится имплантация стимулятора блуждающего нерва и RNS-стимулятора. В первом случае прибор устанавливается подкожно в область ключицы, а его электрод проводят к проходящему в шее блуждающему нерву. По данным проведенных в США и Европе исследований, в 60% случаев данное устройство позволяет снизить количество эпиприступов. Во втором случае прибор вшивается под кожу головы, а его электроды имплантируются в зону эпилептогенного очага. С их помощью, подобно ЭЭГ, устройство постоянно регистрирует электрическую активность мозга. При получении сигналов, свидетельствующих о начинающемся пароксизме, прибор генерирует ответные импульсы, обеспечивающие супрессию эпилептической активности.
Прогноз синдрома Леннокса-Гасто
Синдром Леннокса-Гасто имеет в основном неблагоприятный прогноз. До 10% случаев заканчивается гибелью детей в течение первого десятилетия жизни. Летальные исходы связаны преимущественно с тяжелой травматизацией во время эпиприступов с падением. Прогностически неблагоприятными критериями считаются: манифестация синдрома в более раннем возрасте, начало судорог на фоне ЗПР, предшествующий синдром Веста, высокая частота и интенсивность пароксизмов. Невозможность медикаментозного купирования эпиприступов приводит к прогрессирующей ЗПР. Практически у всех пациентов наблюдается выраженная в различной степени умственная отсталость, половина больных не способны к самообслуживанию.
Источник